Prof. Ing. Roman Martoňák, DrSc. personal web page
room F2-240
What is Computational Materials Science and why we need it?
„The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble. It therefore becomes desirable that approximate practical methods of applying quantum mechanics should be developed, which can lead to an explanation of the main features of complex atomic systems without too much computation.“
P. A. M. Dirac, Quantum mechanics of many-electron systems, Proc. R. Soc. Lond. A 1929 123 714-733
As Dirac wrote in his famous statement nearly 90 years ago, even if we know the microscopic laws, we need a bridge to macroscopic world. The practical realization of this dream is Computational Materials Science, based on numerical approach to quantum mechanics and statistical physics. Employing clever algorithms and powerful hardware we can nowadays, starting from the most fundamental properties of the atoms, predict how they arrange in space and time at given pressure and temperature, eventually forming condensed matter systems, such as crystals and liquids. When we know the structure, we can calculate in principle all mechanical, thermal, electrical, magnetic and optical properties, allowing us to understand a lot of our world and develop practical applications.
Computational Materials Science is a very rapidly developing field of theoretical condensed matter physics. It started in the 50’s and 60’s by introduction of sampling techniques such as Monte Carlo and Molecular Dynamics schemes. Another milestone was the formulation of Density Functional Theory and its combination with finite-temperature statistical mechanics in the form of ab initio molecular dynamics. Due to enormous progress in algorithms as well as in computer hardware over the last decades it gained predictive power and gradually became an invaluable tool in condensed matter physics. It is able to complement the experiment and in some cases even represents the only way to extract information about the properties of the system (e.g. in case of extreme conditions).
Current research directions
-
- study of structure and properties of condensed matter at high pressures
- molecular dynamics and metadynamics-based simulations of structural phase transitions, with focus on nucleation
- structural prediction of crystalline phases
- study of polyamorphism
- development of new methods for computer simulations of condensed matter systems
- study of structure and properties of condensed matter at high pressures
Former research directions
-
- optimization by quantum annealing
- study of quantum and thermal ripples in graphene
- optimization by quantum annealing
Current research grants
APVV-19-0371 Application of machine learning approaches in condensed matter physics (July 2020 – June 2024)
Former research grants
VEGA 1/0640/20 Modeling of new phases and phase transitions in condensed matter (January 2020 – December 2022)
APVV-15-0496 Novel phases and phase transitions in condensed matter (July 2016 – December 2019)
VEGA 1/0904/15 Quantum modelling of structural and electronic properties of solids (January 2015 – December 2017)
APVV-0558-10 Structural and electronic phase transitions on condensed matter systems (May 2011 – August 2014)
APVV-0442-07 Application of dynamical methods in materials science (September 2008 – December 2010)
Media coverage
FMFI UK on our work on graphene english version slovak version
Pravda daily on our work on graphene
Denník N daily on our work on graphene
Interview on Rádio Regina: Hodina s vedou (14 May 2018)
Examples of interesting phenomena in condensed matter from our recent research
A video of the atomic motion during the simulated M1-to-R (Insulator-metal) transition in vanadium dioxide VO2 from our study
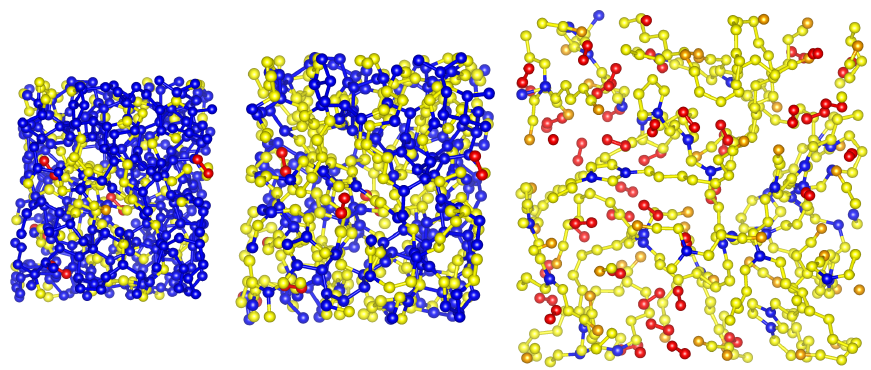
amorphous polymeric nitrogen from our recent study
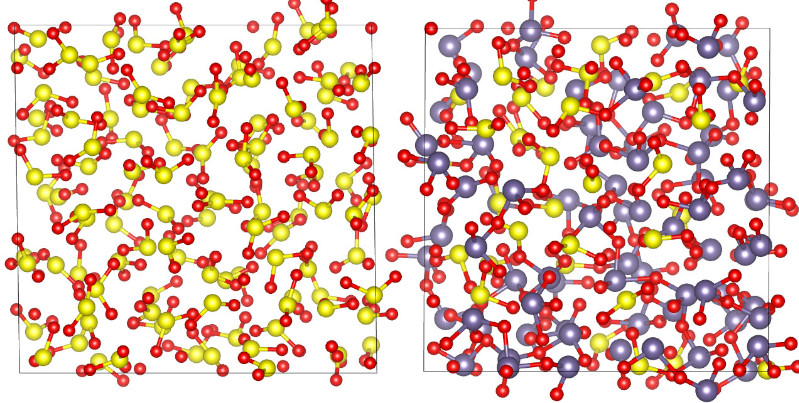
our study of polyamorphism in SO2 (experiment and simulations) was published in PNAS, see popular info (slovenská verzia je tu)
NEWS
very recently our study of nucleation in NaCl was published in Phys. Rev. Lett.

see popular info (slovenská verzia)
Matej Badin is winner of Falling Walls Lab Slovakia 2021 (see info in Slovak)
popular article on high-pressure physics in HPC Focus journal 2022 (Slovak version)